On December 6, 2019, the US Food and Drug Administration
(FDA) gave Amgen its approval
to market the fourth biosimilar infliximab medicine.
Dubbed Avasola (infliximab-axxq), a launch date was not
announced, but one can assume that Amgen intends to launch this agent as soon
as possible. This infliximab biosimilar was approved for all of the autoimmune
indications of the reference product Remicade®.
Avasola will be the third infliximab biosimilar agent to be
marketed, as Pfizer’s Ixifi®, though approved, is not sold in the
US, as it would compete directly with another Pfizer-marketed biosimilar
(Inflectra®). Pricing for the Avasola was not yet available.
Avasola was approved based on
clinical studies involving patients with inflammatory bowel disease, rheumatoid
arthritis, and other indicated conditions (over 4,800 patients taking the drug),
including a phase 3 investigation pitting the biosimilar against the reference
product in patients with rheumatoid arthritis.
The US market for infliximab
is shrinking somewhat, based on falling sales numbers caused by biosimilar
competition and prescriptions lost to other biologics. According to Johnson &
Johnson, parent of Remicade’s manufacturer (Janssen Biotech), US
revenues shrank 24% (to $1.14 billion) in the third quarter of 2019
compared with the same quarter of 2018.
Evidence is mounting, especially from Europe that biosimilars to a single reference product result in the same clinical outcomes, with no appreciable safety issues. Evidence of this type from America will take time to accumulate, because competition in categories with multiple biosimilar options has been much more limited.
The recent announcement in British Columbia of the province’s decision to switch patients with inflammatory bowel disease from Remicade® to biosimilars has once again inflamed some passions. This is part of a broader effort by the Canadian province to increase utilization of biosimilars to gain economic savings. The Canadian GI Society, an advocacy group, published a letter firmly against the plan. They believe only the physician and patient should decide whether a biosimilar switch should occur. The British Columbia government, on the other hand, will pay for either Renflexis® or Inflectra® but not Remicade® after March 5, 2020. Even if patients have been taking the reference product, they will have to switch to one biosimilar or the other.
The Biosimilar Switching Argument
With medical experience supporting the case for switching, the argument by this Canadian advocacy group is less than solid. There may be the need for a medical exception for the infrequent patient taking infliximab, but these drugs can be effectively switched: there is no meaningful difference in effectiveness and safety outcomes on a population-wide basis among infliximab choices.
That said, here is where the supporting arguments get somewhat messy. The Food and Drug Administration (FDA) approves a biosimilar based on its equivalence to a reference product (not to other biosimilars). At the implementation of the Biologics Price Competition and Innovation Act, a biosimilar manufacturer has been required to show that its biosimilar was comparable in physiochemical, pharmacokinetic, and clinical studies to a designated reference product (i.e., US-licensed version of the originator). That required some manufacturers to first prove the equivalence of the US-licensed version to the EU-approved originator in a “bridging study.” This requirement was borne from the understanding that the US- and EU-licensed originator biologics make not be exactly the same either—biosimilars of each other. With more experience, the FDA has backed away from this requirement, understanding that the clinical outcomes differences between two licensed originator products are negligible.
The Laws of Transitivity and Biosimilar Switching
However, the FDA has not revised its opinion on whether two
drugs proved to be biosimilar to one reference product are biosimilar to each
other. Nor whether an interchangeable biosimilar might be considered
interchangeable with other biosimilars. The answer remains: The evaluation
process does not require such testing to gain basic biosimilar approval. In
other words, the evidence did not yet exist.
This all relates to the basic premise of so-called
“nonmedical substitution.” The issue is whether a payer-initiated change with a
noninterchangeable biosimilar is considered to be more like therapeutic or
generic substitution. Therapeutic interchange or substitution takes place when
a drug is switched for another in the same category (e.g., switching Lipitor
for a different statin rather than the generic atorvastatin).
Therapeutic Interchange vs. Biologic Substitution
A payer would be hard pressed to justify an unauthorized switch of adalimumab for infliximab. They are very different TNF-inhibitors. Patient outcomes would likely be affected in some way. Are Inflectra and Renflexis—different infliximab biosimilars—to be considered wholly different biologics? Because the FDA does not endorse that Inflectra and Renflexis are biosimilar to each other, it does imply (whether wrongly or rightly) that they are different biologic brands. If this was considered in the traditional sense, switching might be construed as an example of therapeutic interchange.
Framing the argument in this manner can drive patient and provider resistance to biosimilar uptake. Misinformation? Rather, I believe that this is an embodiment of the ambiguity of our regulatory policies. Certainly, payers don’t subscribe to it; they cover one versus another based more on net costs—certainly not on clinical outcome differences. For payers, it is more black and white; for certain other groups, much remains gray, especially when people believe more decision-making authority is being challenged. Only greater biosimilar clinical utilization, proof of savings, and better dissemination of education about this experience will change advocacy’s perspective .
In a significant coverage move, UnitedHealthcare
(UHC) has signaled that its commercial and Medicaid medical policies on infliximab
and pegfilgrastim have changed direction in favor of the reference drugs.
Effective July 1, 2019, approximately 22.5 million
commercial and 6 million Medicaid UHC members will not be able to access these
biosimilars without trying the reference agents first (virtually eliminating biosimilar
use). Both infliximab and pegfilgrastim are covered generally under the medical
benefit as office-based infusions, and preferring Remicade® and
Neulasta® (including OnPro®).
This move is important for a few reasons. First, it reverses
UHC’s previous position, which preferred the biosimilars over the two
originator products.
Second, it promotes a prior
authorization practice that makes little
sense—since the biosimilar and reference products are expected to work in
the same way and produce similar outcomes, why would a patient who fails Remicade
then be given Renflexis® instead of a different biologic medicine
like adalimumab, ustekinumab, or others?
Third, it implies that both manufacturers have further
reduced the net cost of these drugs to UHC and its customers, undercutting the
current deals offered by the biosimilar manufacturers. If accurate, this is a
positive development in that infliximab and pegfilgrastim prices are continuing
to come down due to competition. It would also indicate that Amgen, maker of
the pegfilgrastim originator Neulasta, is beginning to defend its prefilled
syringe market more aggressively. This is significant, because Amgen had been
more focused on defending the marketshare of its on-body injector (Onpro), which
is dominant. Alternatively, Amgen may be bundling its filgrastim and
pegfilgrastim products more effectively. Coherus
and Mylan had previously announced pricing that would be one-third less
than the list price of Neulasta. Coherus had specifically indicated that it
would be seeking targeted
deals with payers to ensure at least parity position for its prefilled
syringe product Udenyca®. It did not, however, mention UHC as one of
those payers.
Fourth, this move puts a further dent into the
sustainability of the US biosimilar market. Obviously, preferring the
originators will make access to their biosimilars considerably more expensive
for patients. It can only promote greater price cuts by the competing brands
and thus reduce profit margins for the biosimilar manufacturers. In the US, biosimilar
makers need a little encouragement to stay in the market, as very
few have had positive experiences to date (e.g., Pfizer,
Boehringer
Ingelheim, Momenta,
Apobiologix
to name a few).
No one denies the benefits of the increased competition
meaning a halt to price increases and significantly lower net costs, but those
benefits need to be extended across other biologic categories. Without a viable
biosimilar industry, access to lower-cost biologics can only happen through
price controls.
Earlier this month, Pfizer notified the European Medicines Agency (EMA) that it was withdrawing one of its two applications for approval of its biosimilar adalimumab.
According to Pfizer’s Director of Global Media Relations, Thomas Biegi, the company had submitted two applications for this biosimilar, one for a limited set of indications, and the other for the full array of autoimmune indications of the reference product Humira®. Pfizer has decided to focus on gaining approval for the full slate of indications and withdrew the other application. Under the “skinny label,” the product would have been marketed as Fyzoclad™ in Europe. The potential brand name of the biosimilar if approved with all of the reference product’s indications was not disclosed. In the US, the biosimilar is still known as PF-06410293 .
Although Pfizer would not confirm its plans for the US filing, phase 3 trial results for PF-06410293 have been published, establishing the biosimilar’s equivalency to Humira in terms of efficacy, safety, and immunogenicity.
Pfizer noted in its December 5th letter to EMA that their decision was not related to safety or efficacy. No doubt, Pfizer is surveying the heavy competition for adalimumab in Europe today. Pfizer did not elaborate on why the decision was made to submit applications for both the skinny label and the full set of indications.
Pfizer signed a licensing deal with Abbvie on November 30 to market this adalimumab biosimilar in the US. It will be the sixth biosimilar to enter the market in 2023, based on this deal. Therefore, Pfizer must believe that a sixth biosimilar entrant to the US market at that time may still yield relevant revenues and marketshare.
According to EvaluatePharma, Humira US sales estimates (published in 2018) for 2020 will be about $21 billion. By 2024, this company believes Abbvie’s share of the revenue will be a bit more than $12 billion (which is not much different than today’s figures). If this guess is accurate, that leaves $9 billion for seven or so biosimilar makers. If the guess is very inaccurate, and Abbvie is left with far less revenue because of the competition and falling prices, then any number of adalimumab biosimilar manufacturers could attain more than $1 billion in sales.
In other biosimilar news…Amgen has announced the filing of a new biosimilar version of infliximab. ABP 710 was the subject of a phase 3 trial in patients with moderate-to-severe rheumatoid arthritis; researchers concluded that the drug was equivalent to Remicade® in terms of efficacy, safety and immunogenicity. Today’s filing would put this biosimilar on a path to a late Q3 or early Q4 2019 decision by the FDA. If approved, ABP 710 would be the fourth infliximab biosimilar approved in the United States (Pfizer’s Inflixi® is also approved but will only be sold overseas).
This post was updated and corrected on December 18, 2018.
Payers have not been quick to add biosimilar infliximab to their drug coverage. Yet, biosimilar switching is the objective for most health plans and insurers who are thinking about long-term savings. Even if they do not exclude the reference product Remicade® from coverage, some health plans, like Kaiser, have been moving forward in this effort.
At the Academy of Managed Care Pharmacy’s Nexus 2018 meeting in Orlando this week, two clinical pharmacy specialists from Kaiser Foundation Health Plan of the Northwest described what may be a best practice in converting patients to biosimilar Inflectra®.
RELEVANCE OF BIOSIMILAR SWITCHING AT THE PLAN LEVEL
Kayla Hubrich, PharmD, emphasized the importance of patient education, and patients’ reliance on Google for research. She said, “When patients will turn to Google and type in ‘Should I switch to an infliximab biosimilar?’ the first search result they see is an ad for ‘Finely Tuned,’ a Janssen website.” This, of course, discourages the use of biosimilars.
At Kaiser Foundation Health Plans, coverage decisions are made at a national level for its 12.5 million members and implemented at the regional plan level, according to Lynsey Smith, PharmD. The health plan made Zarxio® its preferred filgrastim product in 2016, and registered 96% of all filgrastim dispensings in self-injected settings, and 100% of all clinical administrations for this biosimilar.
Source: Kaiser Foundation Health Plan
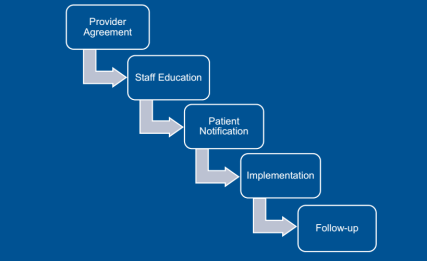
Obtaining that level of use means that not only treatment-naive patients were using Inflectra, but also those using Remicade in the past. Dr. Smith outlined the key steps in this conversion, starting with the providers. “For new starts,” said Dr. Smith, “the tactic was just to have the doctor choose the biosimilar” using tools incorporated into the electronic health record that encouraged them to order the preferred product. Concerning those patients needing to be converted from the reference product, Kaiser asked the prescriber to sign a ‘Therapeutic Equivalency Protocol’ agreement, which authorized the plan to make the switch. The biosimilar switching agreement was voluntary, and virtually all the rheumatologists, dermatologists, and gastroenterologists signed. “One GI out of 20 declined to authorize the switch in patients already receiving Remicade,” she said.
Kaiser emphasized patient notification and education. A letter, signed in their doctor’s name, was sent to each patient at least 2 weeks before the conversion date, explained Dr. Smith. Clinical Pharmacy Services was enlisted to answer patients’ questions via phone and E-mail. Patients were also given informational handouts about the biosimilar switching program at their infusion center.
“During this process, the clinical pharmacists received 30 to 40 calls,” she said. “The patients’ main concerns were whether the product was going to work as well as their old drug and whether they would receive the same copay assistance as before.” Active patient outreach was not conducted after the switch was instituted. Any patients reporting issues or concerns were triaged through Clinical Pharmacy Services.
Dr. Hubrich added that infusion center pharmacists reviewed all patients scheduled for infusions one week ahead of their appointment. The infusion center confirmed that the provider signed a TEP document, that patients were sent the notification letter, and that the infliximab order changed to Inflectra. Kaiser also developed a nurses’ protocol for the biosimilar switch and worked to educate practice staff about the program.
INFLIXIMAB SWITCHING PROGRAM RESULTS
The conversation program began on May 1, 2017, with dermatologists and rheumatologists, focusing on patients who were getting their first infliximab treatment. Dr. Hubrich stated that notification letters were sent to 158 patients. Three weeks later, current patients began to be switched from Remicade to Inflectra. The GI conversion began on May 11, 2017 with treatment-naïve patients, and letters were sent to 188 adult patients (as Inflectra did not have the pediatric ulcerative colitis indication). Active therapeutic switching began in September. “The one GI who declined to sign the TEP agreement joined in 2018,” said Dr. Hubrich. This is likely because of the experience of this doctor’s peers.
A total of 22 patients (6.4%) across specialties reported adverse events, with nine being changed back to reference product (2.6%), five changed to a different medication class, four resulted in a dosage increase, one patient decided to discontinue therapy, and three continuing biosimilar infliximab treatment without any change. They did find that 12.8% of patients experienced some “nocebo” effects, despite the fact that “no statistically significant changes in effectiveness and safety were observed after a medican of four infusions in 9 months of study.”
Dr. Smith asserted that communication was critical to the success of the program, with patients and providers. The provider’s agreement to sign the TEP document was a necessary step, and was accepted by all Kaiser’s specialist providers.
It must be emphasized that Kaiser has a different magnitude of leverage over its physicians than a network plan like Aetna or CIGNA. Yet a biosimilar switching program like this could be a blueprint for other integrated health plans to move forward if they desire to move patients quickly and efficiently to biosimilar therapy.
The Centers for Medicare and Medicaid Services (CMS) has decided drugs covered under Medicare part B may be subject to step therapy, if so desired by Medicare Advantage plans. UnitedHealthcare has become the first to publicly implement step therapy policies for these drugs. However, biosimilar step therapy is not the typical utilization management tool that industry executives are used to seeing.
Traditional step therapy or step edits for prior authorization policies are typically used to require the use of an effective, low-cost drug class before trying a more-expensive treatment. For example, a plan might have a step in place before a patient can receive Humira®, such as requiring documented failure on other disease-modifying anti-rheumatic drugs, like azathioprine or methotrexate. This makes very good sense when supported by practice guidelines or treatment pathways, based on solid supportive evidence.
For biosimilar manufacturers, the perspective on the revised CMS policy, seems to imply trying the biosimilar before receiving the branded originator product. This biosimilar step therapy would make very little sense. A doctor would not be practicing evidence-based medicine if he or she prescribed Remicade® to a patient after failure of Renflexis®. There is no evidence to show that the biosimilar will work in a patient who did not receive adequate clinical benefit from the reference product (and vice versa). Similarly, there is no information to show that a patient who has an adverse effect while taking Remicade will not have that adverse effect after injecting with Renflexis (or vice versa). In other words, after failing one, a new mechanism of action should be tried, not a product with a very similar structure. This may be a different argument, if a subcutaneous form of infliximab was introduced, and this might be reason to step the infusible form through this drug.
In United’s announcement, they are clearly seeking to increase biosimilar utilization, as designated preferred part B agents, at the expense of Remicade use, the nonpreferred agent. Therefore, it may make more sense that new patients will have to use a biosimilar before being prescribed the reference product. Step therapy in this case is almost an aside.
Ironically, the Department of Health and Human Services has also expressed its desire to move part B agents like self-administered injectables to part D. This may not apply to infliximab, as it is given as an in-office infusion. Should this be the case, plans will have many pharmacy tools at their disposal beyond biosimilar step therapy.
In other biosimilar news…Fresenius Kabi has signed an agreement with Abbvie to delay its adalimumab biosimilar market entry in the US until 2023. The manufacturer is currently trying to secure European approval for the product. A 351(k) application has not yet been filed by Fresenius in the US.
When Pfizer first announced its lawsuit against Janssen’s parent Johnson & Johnson in September 2017, it pointed to exclusionary contracting, “anticompetitive” behavior of Remicade®’s maker as the reason for its very limited market access.
The lawsuit claimed that Janssen has withheld or threatened to withhold rebates if payers do not keep Remicade in an exclusive preferred position. The degree to which health plans knuckled under to these demands may only be inferred from the 3% marketshare Pfizer’s Inflectra® now holds. For these drugs, which are still typically covered under the medical benefit, “nonpreferred positioning” usually means no coverage. For drugs covered under the pharmacy benefit, this is not the case.
In August, the Eastern Court of Pennsylvania ruled against J&J in its request that the lawsuit be dismissed. While discovery in the case may be ongoing, we could not find mention of a resolution date for the suit.
For the sake of argument, let’s say that the Eastern Court of Pennsylvania rules in favor of Janssen. In other words, exclusionary contracting was not an anticompetitive behavior. That means the status quo is intact, but some factors may affect this situation going forward. These include the Center for Medicare and Medicaid Services’ desire to move part B drugs (the medical benefit) to part D (the pharmacy benefit) for Medicare beneficiaries.
The scrutiny on rebate contracting coming from several sectors, and lack of transparency, may also independently influence future use of these pharmaceutical company tactics. I helped conduct a market research project recently on a nonspecialty drug. As part of these interviews, we were asked by the client to inquire about the range of rebates they were receiving from competitor manufacturers. Their responses were requested as a range (e.g., 20% to 30%), not specific contract details, and we had no intention of providing reports of individual payer deals, only anonymous, aggregate information. We expected little to no response to that query, and that is exactly what we received.
Let’s discuss the other potential outcome, in which the Court rules in favor of Pfizer. That implies that this exclusionary contracting practice is indeed anticompetitive. If this is the case, we may be on a very slippery slope. What is the difference between payers and pharma companies engaging in a “1 of 1” contract when there are multiple potential products and a “1 of 2” contract? In both cases, drug makers are committing payers to anticompetitive behavior (as perhaps defined by the Court’s new precedent).
The preferred drug tier (whether preferred generics, preferred brands or whatever) is supposed to be for products with proven clinical, patient care, or economic advantages. Truthfully, payers rarely place medications in the preferred tier for reasons other than net costs or rebate contracting, which is based on marketshare.
Now add in the potential effects of the Administration’s desired shift to part D, where pharmacy benefit rules can be applied. That exposes injectable products that were shielded under Medicare part B to commonly applied formulary placement practices.
To be complete, Janssen’s strategy was not solely based on Remicade. It may be found to have bundled Remicade with other agents in deals to exclude Pfizer’s products. The Court may also react specifically to Janssen’s contract stipulation that threatens to withhold rebates connected to future use of the product, to increase its leverage.
However, if the Court determines that 1 of 1 or exclusionary contracting with rebates are the root of the anticompetitive behavior, why should 1 of 2 or even 1 of 3 contracts in a drug category with 5 similar agents be less so? This is the slippery slope that could undo rebate contracting, and push us towards a system that more resembles a competitive bidding process like in Europe. Alternatively, it could accelerate the move towards outcomes- and value-based contracting. The result could be a system-wide revamping of the drug formulary and the pharmacy–drug maker relationship.
In other biosimilar news…Sandoz has signed a licensing agreement with Abbvie, allowing it to market its biosimilar version of Humira in 2023. The agreement, as with Abbvie’s settlements with other biosimilar makers, halts all patent litigation with Sandoz in exchange for a licensing royalty paid to Abbvie.
Everyone with an opinion believes that biosimilar drug use will save the health system considerable money. Calculations for biosimilar savings have been hampered by several factors. For example, previous high estimates have not been based on real-life scenarios. Only 3 biosimilars have been launched and utilized in the US; so little experience has been gained on which to base calculations.
Yet, isolating the savings associated with a single approved biosimilar does put their potential into perspective. It also demonstrates the promise of cumulative biosimilar savings with their launch and uptake. Based on current infliximab average sales prices (ASPs), which considers discounts and rebates, one organization believes that a 50% marketshare for biosimilar infliximab could result in well over $400 million in annual savings system wide.
The analysis, conducted by Wayne H. Winegarden, PhD, Senior Fellow in Business and Economics, Pacific Research Institute, accrued the lion’s share of the annual savings to employer-sponsored health plans ($262 million to $315 million, compared with no sales of infliximab biosimilars). Medicare accounted for up to $150 million savings annually.
Dr. Winegarden tested several scenarios. The calculation considered the cost of the infliximab regimen based its various indications. He calculated biosimilar savings using different add-on percentages to ASP (including the current ASP + 4.3% payment and up to ASP + 20%), as well as different marketshares of the biosimilars (from 10% to 90%).
The current marketshare of the two available infliximab biosimilars—Inflectra® and Renflexis®—is below 5%, based on data from the first quarter of this year. This is partly because of Janssen’s tactics in matching the net costs of biosimilars with additional rebates on Remicade. This raises two important points: Dr. Winegarden’s analysis reveals savings accruing to the health care system (not necessarily to the payer). Also, the very existence of infliximab biosimilars has resulted in significant net savings compared with the price increases seen prior to their introduction.
It is a bit more difficult to pinpoint the system savings resulting from the use of the first biosimilar approved in the US, filgrastim-sndz (Zarxio®). The other branded product, tbo-filgrastim (Granix®), was launched a couple of years earlier and gained its own marketshare from the reference brand Neupogen®. No doubt, Zarxio contributed to some level of cost savings. In other words, the infliximab example is an easier calculation with a cleaner result.
With eight biosimilars for six reference products awaiting their turn to hit the market, and drugs like adalimumab and etanercept among them, it is easy to see how biosimilars savings can easily exceed $10 billion. Just not yet.
When payers, patients, or physicians discuss biosimilars, they assume that the biosimilar works just like the reference product. They also assume that the biosimilar is administered in the same way as the originator biologic. Celltrion is actively researching a new subcutaneous infliximab. This could result in a first for the biosimilar industry.
Sponsored by Celltrion and conducted in multiple sites, the research results were announced at the annual meeting of the European Congress of Rheumatology in June. The investigators presented outcomes data on the use of a subcutaneous (SC) form of infliximab-dyyb. Currently, infliximab is only available as an intravenous (IV) infusion at the physician’s office that takes at least 2 hours. Subcutaneous infliximab was given on a biweekly basis.
The researchers studied 48 patients with rheumatoid arthritis, finding that outcomes were not clinically different through 30 weeks of follow-up. Three dosages were tested, and in this small study, no ACR20 differences were reported in any subgroup receiving infliximab infusions or SC injections.
Hypersensitivity reactions did occur in one patient each receiving the lowest dose (90 mg) SC and the middle dose (120 mg). None were seen in the group receiving the highest infliximab SC dose (180 mg). Injection site reactions occurred in two patients apiece in the 90 mg and 180 mg dose cohorts. receiving subcutaneous infliximab. The formation of antidrug antibodies was detected in nine patients receiving the standard infusion, but less than half that number in each of the subcutaneous groups.
Currently, infliximab treatment requires a lengthy office visit for each infusion (every 8 wk in the maintenance phase). It is one of the key limiting factors to its use. A self-injectable formulation should result in lower administration costs, and the potential for covering the agent through the pharmacy benefit.
A phase 1, open-label trial of subcutaneous infliximab has already been conducted by Celltrion in patients with Crohn’s disease. That trial found similar outcomes between the SC and IV formulations. Another phase 1 trial is wrapping up, this one evaluating safety and pharmacokinetics in healthy volunteers. Celltrion is also sponsoring a phase 3 trial of more than 300 patients with rheumatoid arthritis. Preliminary results will not be available until December 2018.
It is not yet clear, however, what type of data the Food and Drug Administration would require for approval of a new formulation of a biosimilar. The regulatory agency may decide to treat this as it would a new route of administration for any approved product, which would focus on pharmacokinetic and pharmacology factors. Celltrion seems to be covering all of its bases.
Health plans and insurers are not yet turning to biosimilar infliximab as a preferred therapy, according to Gillian Woollett, DPhil, MA, of Avalere. Her new report surveyed publicly available policy about health plans across the nation. The principal finding was that step therapy was commonly used to encourage use of the originator product.
In fact, just one health plan (representing 1% of the 172 million lives covered in this study) supported the use of either Inflectra® or Renflexis® over the reference product Remicade® through step therapy. One plan (2% of the covered lives) allowed the use of either the originator product or Inflectra as a first step.
Gillian Woollett
Four of the 18 plans with publicly available information did not utilize step-therapy rules for any forms of infliximab. However, “10 of the 18 plans (55% of plans, 52% of covered lives) require the use of [Remicade] first, alone or in combination with another DMARD,” stated Dr. Woollett in the report. A total of 81% of the covered lives from these 18 plans were subject to step therapies limiting access to one infliximab product or the other.
On its face, this type of step policy makes a bit of sense. Step therapies are often used alone or part of prior authorization mechanisms to make sure patients try more cost-effective agents first. In rheumatoid arthritis, that may comprise use of nonbiologic drugs before proceeding to a TNF inhibitor and then to another biologic in patients with rheumatoid arthritis. However, there is no proven benefit (or even logic) to offering a biosimilar infliximab after failing Remicade, or vice versa. If there was a significant clinically relevant difference in immunogenicity, this could be an issue, but this also has not been seen in practice. It makes more sense to try another anti-TNF or perhaps even move to an interleukin inhibitor—something with a different (or slightly different) mode of action.
A policy such as this can confuse the issue for patients, whose knowledge of biosimilars seems tenuous, and even providers, some of whom have little experience prescribing them, particularly because of payers’ Remicade-first policies.
The Avalere report provides some support for how payers are arresting utilization of biosimilar infliximab in favor of the originator infliximab product.
Dr. Woollett paints a very different picture for subcutaneously administered filgrastim products. Forty-nine percent of the covered lives (five large plans) had policies favoring Zarxio®, whereas 27% of covered lives were encouraged to use Neupogen® first. For these 18 plans, five (28% of plans, 49% of covered lives) demonstrate a preference for the biosimilar, filgrastim-sndz. Five (28% of plans, 27% of covered lives) demonstrate a preference for the reference filgrastim. Eight plans (44% of plans, 24% of covered lives) do not indicate a preference through formulary design. A further 24% were not subject to any preference.


 Traditional step therapy or step edits for prior authorization policies are typically used to require the use of an effective, low-cost drug class before trying a more-expensive treatment. For example, a plan might have a step in place before a patient can receive Humira®, such as requiring documented failure on other disease-modifying anti-rheumatic drugs, like azathioprine or methotrexate. This makes very good sense when supported by practice guidelines or treatment pathways, based on solid supportive evidence.
Traditional step therapy or step edits for prior authorization policies are typically used to require the use of an effective, low-cost drug class before trying a more-expensive treatment. For example, a plan might have a step in place before a patient can receive Humira®, such as requiring documented failure on other disease-modifying anti-rheumatic drugs, like azathioprine or methotrexate. This makes very good sense when supported by practice guidelines or treatment pathways, based on solid supportive evidence.
 exactly what we received.
exactly what we received. The researchers studied 48 patients with rheumatoid arthritis, finding that outcomes were not clinically different through 30 weeks of follow-up. Three dosages were tested, and in this small study, no ACR20 differences were reported in any subgroup receiving infliximab infusions or SC injections.
The researchers studied 48 patients with rheumatoid arthritis, finding that outcomes were not clinically different through 30 weeks of follow-up. Three dosages were tested, and in this small study, no ACR20 differences were reported in any subgroup receiving infliximab infusions or SC injections.