Just a few short weeks ago, Abbvie announced that it intended to rely on discounts as deep as 80% in parts of the EU to retain Humira® marketshare. One bellweather EU member country has signaled that it is signing tenders with other biosimilar adalimumab manufacturers.
The Center for Biosimilars reported an Email exchange with the Danish national tendering authority Amgros, which manages the country’s bidding system. Amgros confirmed that Abbvie did not provide the best bid for two tenders for adalimumab (covering January to March 2019 and covering April to December 2019). Five companies (including Abbvie) competed for the national tenders. Although Abbvie did not rank best for pricing, agreements were signed with all five companies.
According to the report, a spokesperson for Amgro said, “In both tenders for adalimumab 40 mg, we have entered into agreements with 5 companies—the agreements are ranked according to price. In both tenders, we have signed an agreement with Abbvie for Humira—but Humira does not have the lowest price (ie, is not the winner with the highest ranking).”
The importance of this action may extend beyond Denmark, as several European countries utilize others’ pricing decisions as a benchmark for their own. For example, the price for adalimumab in Bulgaria by policy cannot exceed that in 17 other EU countries.
Journalist Dan Stanton reported that Amgen withdrew in September from The Biosimilars Forum, based on disagreements with the remaining eight biosimilar-manufacturing members and perhaps internal conflicts within Amgen.
Founded by 11 members (Allergan, Amgen, Boehringer Ingelheim, Coherus BioSciences, EMD Serono, Epirus Biopharmaceuticals, Merck and Co., Pfizer, Samsung Bioepis, Sandoz, and Teva), 8 now remain (B-I, Coherus, Fresenius Kabi, Merck, Pfizer, Samsung Bioepis, Sandoz, and Teva).
An Amgen spokesperson told Mr. Stanton, “As one of its founding members, Amgen supports the Forum’s mission to advance biosimilars and improve access to biological medicines. Although aligned on this mission, Amgen and the Forum disagree on how best to support the establishment and growth of a vibrant US biosimilars market.”
Areas of disagreement may have arose over the need for policies to support widespread acceptance of biosimilars and innovation in originator biologics, and the types of education and how it is disseminated to support uptake.
Amgen harbors a healthy pipeline of biosimilars as well as defending its brands against biosimilar competition. Whereas its Epogen®, Neupogen®, and Neulasta® are under active assault by biosimilars, its biosimilar versions of adalimumab (Amjevita®) and bevacizumab (Avastin®) are both approved but not marketed in the US.
As was pointed out by Mr. Stanton’s report, Amgen sponsored a YouTube video that supported use of naming conventions that differentate biosimilars from reference products (against the Biosimilar Forum’s advocacy) as well as implying that biosimilar switches can result in negative outcomes. Earlier in November, Forum-member Sandoz issued a statement in support of Pfizer’s Citizen’s Petition, complaining of inaccurate and misleading statements made by makers of reference biologics.
Amgen (as well as Pfizer and Boehringer Ingelheim) must walk a tightrope that other biosimilar-focused manufacturers do not. To be leading innovative drug makers, they systemize their efforts to research new medicines and acquire drug discovery firms, engage in lifecycle management, and aggressively protect their intellectual property. Yet both drug makers seem committed to the biosimilar side of their pipeline and growing the value of their biosimilar enterprises.
The Biosimilars Forum, formed in 2015, is an advocacy organization, competing in the policy space with the Biosimilar Council, which has members that represent a more diverse group (Apobiologix, AmerisourceBergen, Amneal Biosciences, Axinn, Biocon, Biorasi, Boehringer Ingelheim, Dr. Reddy’s Laboratories, Lupin, Momenta Pharmaceuticals, Mylan, and Sandoz). The Biosimilars Council is a division of the Association for Accessible Medicines.
Coherus Biosciences surprised many on its third-quarter earnings call late yesterday. It will rely not on a lower price than its biosimilar competitor to gain marketshare after Coherus’ Udenyca launch, but on its ability to pull through on its patient and provider services and supply chain to gain significant marketshare for its biosimilar version of Neulasta®.
This is not to imply that Coherus will not offer contracts to group purchasing organizations (GPOs), hospitals, and payers. The company intends to do so. However, the wholesale acquisition cost (WAC) for Udenyca® will match that of Mylan’s Fulphila®—$4,175 per vial, or a 33% discount from Amgen’s reference product. Denny Lanfear, CEO of Coherus added that the company’s contracting plans “will deliver additional value to payers.”
Jim Hassard, Coherus
AWAITING HCPCS CODING
Unlike other biosimilar manufacturers, this is their first product to reach the market. Not only was manufacturing and production a priority, but company infrastructure had to be ready for launch. Although Coherus pointed out that the sales force for Coherus is fully in place, they are holding back the Udenyca launch until the Center for Medicare and Medicaid Services (CMS) designates a Q code for claims and billing purposes. Therefore, the goal is a Udenyca launch date of January 3, 2019.
Jim Hassard, Vice President for Marketing and Market Access, emphasized that “Our overall launch strategy goes beyond pricing, to reliable supply and services. We’re committed to world-class execution and salesforce effectiveness.” The company’s Coherus Complete, patient and provider service site, is operational, and this will include copay support for eligible patients. Mr. Hassard stated, “This price is attractive to payers without diminishing our value proposition. We can deliver significant savings to the health system versus Neulasta.”
CAN UDENYCA GRAB SOME ONPRO MARKETSHARE?
One interesting statement made during the call was the expectation that Coherus will go after some of Neulasta Onpro’s share of the market. Amgen’s on-body injector accounts for about 60% of all Neulasta utilization today, “but this growth has flattened out,” Chris Thompson, Vice President of Sales, emphasized. “We’re looking at the whole market, not just prefilled syringe market,” he said. “We think we’ll be able to sell through the Onpro market,” meaning that their pricing and services will attract some of this marketshare. In fact, Coherus executives believe that biosimilars may eventually garner nearly 70% of the pegfilgrastim market.
Coherus believes that there is pent-up demand for the biosimilar in the hospital segment today, which is why GPOs may represent promising contracting opportunities. They are seeking parity positioning at the payer and pharmacy benefit manager level.
This sounds fairly reasonable. Yet the vast majority of biosimilar consultants and payers with whom I had communicated had anticipated that Coherus would launch with at least a modest WAC discount relative to Mylan’s Fulphila. On the conference call, the investment banking participants wanting information on the Udenyca launch seemed caught off guard as well.
UDENYCA REVENUE TO SUPPORT COHERUS FOR NOW
Perhaps this strategy gives Coherus ample room for contracting while retaining a respectable net cost. Mr. Thompson said, “We’ll roll out a comprehensive contracting strategy for GPOs in the next week or two. It will be competitive and designed to win.”
It may need to be. Relying on better services and perhaps even a better supply chain (albeit one that is brand new) may not be sufficiently persuasive to hospital and payer P&T Committees. And Coherus needs to generate revenue from its sole product to feed its new sales team, new product development, and hungry investors.
The Food and Drug Administration (FDA) announced yesterday the approval of adalimumab-adaz from Sandoz. The new agent, dubbed Hyrimoz™, will not be launched in the US until 2023. The approval of Hyrimoz is the third for Sandoz (but only one, Zarxio®, is available for prescription in the US).
The FDA approval of adalimumab-adaz covered several indications, including adult Crohn’s disease, ankylosing spondylitis, juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, rheumatoid arthritis, and ulcerative colitis. The drug’s approval was based partly on the findings of a phase 3 clinical trial in patients with chronic plaque psoriasis, in which the biosimilar was found to be noninferior to the originator product Humira® in terms of efficacy (i.e., PASI 75 score) and safety.
Hyrimoz is the third approved adalimumab biosimilar, none of which have been marketed due to patent litigation. Abbvie has signed licensing agreements with Amgen and Samsung Bioepis to delay US launches.
HUMIRA PRICE DISCOUNT IN THE EU
This biosimilar is being marketed in the EU, competing with several others for the Humira marketshare overseas. However, signs of real competition are heating up in Europe, as Abbvie has offered a Humira price discount of as much as 80%.
According to an article published in Fierce Pharma, Abbvie is hoping to squash the biosimilar competition and prevent it from gaining valuable European experience ahead of US launches in 2023. The article cited a report by Bernstein analyst Ronny Gal, indicating that even at an 80% discount, Humira will still be profitable for Abbvie. “The objective is to defend the US market by denying the biosimilars in-market experience [in Europe] and then arguing the Europeans ‘chose’ Humira over the biosimilars for quality reasons beyond price,” according to Gal’s report.
On the other hand, this puts the biosimilar makers in a tight spot on the continent. They need to earn back their R&D costs and may be unwilling to face an immediate low-profit reality. Revenues within the EU for Humira are $4 billion. Even if it offered tenders of 80% for every member country (and they were accepted), revenues would still be in the range of $800 million. This would drastically reduce the size of the revenue slices for the European biosimilar competitors. It could be possible that some may drop out of the market, at least until the time of the US launches.
Payers have not been quick to add biosimilar infliximab to their drug coverage. Yet, biosimilar switching is the objective for most health plans and insurers who are thinking about long-term savings. Even if they do not exclude the reference product Remicade® from coverage, some health plans, like Kaiser, have been moving forward in this effort.
At the Academy of Managed Care Pharmacy’s Nexus 2018 meeting in Orlando this week, two clinical pharmacy specialists from Kaiser Foundation Health Plan of the Northwest described what may be a best practice in converting patients to biosimilar Inflectra®.
RELEVANCE OF BIOSIMILAR SWITCHING AT THE PLAN LEVEL
Kayla Hubrich, PharmD, emphasized the importance of patient education, and patients’ reliance on Google for research. She said, “When patients will turn to Google and type in ‘Should I switch to an infliximab biosimilar?’ the first search result they see is an ad for ‘Finely Tuned,’ a Janssen website.” This, of course, discourages the use of biosimilars.
At Kaiser Foundation Health Plans, coverage decisions are made at a national level for its 12.5 million members and implemented at the regional plan level, according to Lynsey Smith, PharmD. The health plan made Zarxio® its preferred filgrastim product in 2016, and registered 96% of all filgrastim dispensings in self-injected settings, and 100% of all clinical administrations for this biosimilar.
Source: Kaiser Foundation Health Plan
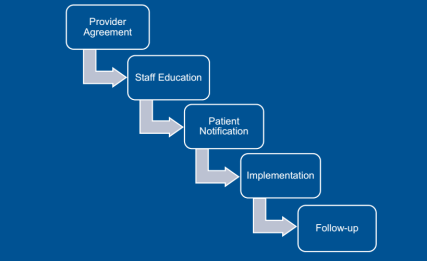
Obtaining that level of use means that not only treatment-naive patients were using Inflectra, but also those using Remicade in the past. Dr. Smith outlined the key steps in this conversion, starting with the providers. “For new starts,” said Dr. Smith, “the tactic was just to have the doctor choose the biosimilar” using tools incorporated into the electronic health record that encouraged them to order the preferred product. Concerning those patients needing to be converted from the reference product, Kaiser asked the prescriber to sign a ‘Therapeutic Equivalency Protocol’ agreement, which authorized the plan to make the switch. The biosimilar switching agreement was voluntary, and virtually all the rheumatologists, dermatologists, and gastroenterologists signed. “One GI out of 20 declined to authorize the switch in patients already receiving Remicade,” she said.
Kaiser emphasized patient notification and education. A letter, signed in their doctor’s name, was sent to each patient at least 2 weeks before the conversion date, explained Dr. Smith. Clinical Pharmacy Services was enlisted to answer patients’ questions via phone and E-mail. Patients were also given informational handouts about the biosimilar switching program at their infusion center.
“During this process, the clinical pharmacists received 30 to 40 calls,” she said. “The patients’ main concerns were whether the product was going to work as well as their old drug and whether they would receive the same copay assistance as before.” Active patient outreach was not conducted after the switch was instituted. Any patients reporting issues or concerns were triaged through Clinical Pharmacy Services.
Dr. Hubrich added that infusion center pharmacists reviewed all patients scheduled for infusions one week ahead of their appointment. The infusion center confirmed that the provider signed a TEP document, that patients were sent the notification letter, and that the infliximab order changed to Inflectra. Kaiser also developed a nurses’ protocol for the biosimilar switch and worked to educate practice staff about the program.
INFLIXIMAB SWITCHING PROGRAM RESULTS
The conversation program began on May 1, 2017, with dermatologists and rheumatologists, focusing on patients who were getting their first infliximab treatment. Dr. Hubrich stated that notification letters were sent to 158 patients. Three weeks later, current patients began to be switched from Remicade to Inflectra. The GI conversion began on May 11, 2017 with treatment-naïve patients, and letters were sent to 188 adult patients (as Inflectra did not have the pediatric ulcerative colitis indication). Active therapeutic switching began in September. “The one GI who declined to sign the TEP agreement joined in 2018,” said Dr. Hubrich. This is likely because of the experience of this doctor’s peers.
A total of 22 patients (6.4%) across specialties reported adverse events, with nine being changed back to reference product (2.6%), five changed to a different medication class, four resulted in a dosage increase, one patient decided to discontinue therapy, and three continuing biosimilar infliximab treatment without any change. They did find that 12.8% of patients experienced some “nocebo” effects, despite the fact that “no statistically significant changes in effectiveness and safety were observed after a medican of four infusions in 9 months of study.”
Dr. Smith asserted that communication was critical to the success of the program, with patients and providers. The provider’s agreement to sign the TEP document was a necessary step, and was accepted by all Kaiser’s specialist providers.
It must be emphasized that Kaiser has a different magnitude of leverage over its physicians than a network plan like Aetna or CIGNA. Yet a biosimilar switching program like this could be a blueprint for other integrated health plans to move forward if they desire to move patients quickly and efficiently to biosimilar therapy.
In part two and the conclusion of this interview, Molly Burich, MS, Director, Public Policy: Biosimilars and Pipeline, speaks to Boehringer Ingelheim’s progress in Cytelzo interchangeability studies, its plans for the product in Europe in the face of several adalimumab biosimilars launches in the EU, and also the complexity inherent in CMS’s plans to move biologic agents from part B to part D coverage.
Molly Burich, MS
BR&R: Boehringer Ingelheim indicated that it started the study on Cytelzo interchangeability last year. What’s the progress on this effort?
Burich: The trial is continuing to progress. It’s a high bar and a big commitment. We will certainly publicize relevant information in due course.
We feel that for Cyltezo, in particular, interchangeability is an important component. It may drive switching. The study will also show a complement of clinical data around that topic. We hope to have information to share in the future. [Editor’s Note: The VOLTAIRE-X study, which will evaluate the effect of switching between Cyltezo and Humira in patients with plaque psoriasis, has an estimated primary study completion date of March 2020 and full study completion of July 2020, according to ClinicalTrials.gov]
BR&R: Speaking about Cyltezo, I have a question about the marketing floodgates being opened in the EU for adalimumab biosimilars. At least 4 are being launched in the EU after the October 16th patent expiration. Does Boehringer Ingelheim plan to join the fray?
Burich: Boehringer Ingelheim had planned to bring Cyltezo to patients in the EU. Due to the patent litigation with AbbVie in the US, we will not commercialize Cyltezo in the EU. Boehringer Ingelheim will continue all activities for our biosimilar in the United States. We are committed to making Cyltezo® available to U.S. patients as soon as possible and certainly before 2023.
PART B TO PART D TRANSITION BY CMS
BR&R: Medicare has indicated that it will move many Medicare part B drugs into part D. To what extent will this affect biosimilar access and utilization?
Burich: It is a very hot topic these days. We have some pretty significant concerns on conceptually around what it means for moving from part B to part D. The key reason revolves around the access question, including patient cost sharing.
A move from part B to plan D could mean that patient cost sharing may jump significantly. We know that part B beneficiaries have wraparound or Medigap coverage to protect them from cost sharing issues. In part D, there is not such protection. Aside from the biosimilar question, the move from part B to part D really has to be explored and discussed a lot more to understand how we can ensure that patient access is not reduced through high cost sharing. That needs to be ironed out as it applies to any part B drug before we can speculate whether this is an opportunity for a biosimilar. Time will tell what that really looks like.
Last month, CMS released the Medicare Advantage guidance allowing for step therapy for part B drugs. That could be a potential opportunity for biosimilars, if we know how some of the access concerns will be addressed. We just don’t have the full picture at this point.
BR&R: Is it possible that this move to part D might spur some payers to create biosimilar tiers? These would require lower cost sharing for patients compared with reference biologics, assuming contracts with the reference manufacturer permits it.
Burich: In my opinion, we’ll need access to more biosimilars before we see a lot of that activity. It’s hard to foresee what big benefit design changes will be coming, but it’s certainly possible. We’ll need a mature market in the US before that will happen.
BR&R: The devil is in the details with this switching issue but there’s also an access issue. Plans can make midyear formulary changes, this would then apply to biosimilars and reference drugs covered under part D.
Burich: This is an important issue. The latest guidance that we saw from CMS, which is now a couple of years old, allowed positive formulary changes. Adding the biosimilar to a formulary is always allowed mid-year. The question involves removing an originator product or changing its tier.
CMS has said that those situations would be reviewed on a case-by-case basis. These rules preventing negative formulary changes midyear are there to protect patient access. It will take CMS some time to iron out what the process looks like for this type of potential formulary change midyear. For now, we’ll have to rely on CMS’s case-by-case review
BR&R: In general, payers do not consistently fund and manage self-injectable specialty drugs in the same way. In some cases, they cover these agents under the pharmacy benefits, medical benefit, or even both. Further, they can be managed under either benefit as well. However, it seems we are moving toward pharmacy management of these agents. How does this affect biosimilar access, if at all?
Burich: There will be more benefit design changes once we have a more robust biosimilar market. More specifically, when we have pharmacy benefit biosimilars.
We’ve mentioned CMS’s intention to move more of these products from part B to part D. It is possible that commercial plans will have different benefit designs and treat injectables differently than Medicare does. We want to make sure that biosimilar or not, the access piece is really at the center of those changes; it will not be beneficial to the biosimilar market if this move causes significant patient access issues (e.g., actual access to this drug or big swings in cost sharing). All of those things will be equally problematic for a biosimilar as they are for an originator, so we want to make sure we have our eye on the access component.
BR&R: Health and Human Services Secretary Azar and FDA Commissioner Gottlieb have loudly stated their desire to improve biosimilar patient and market access. The Biosimilar Action Plan was released earlier in the summer to that end. What is the one aspect of the Biosimilar Action Plan that appeals most to manufacturers like Boehringer Ingelheim?
Burich: The aspect of education, tackling both proactive education and countering misinformation is very critical from our perspective. We’d like to see more materials moving forward that focus on switching and on interchangeability. We haven’t really scratched the surface on those topics from an education standpoint.
The reality is that the FDA has an important voice and bringing validity to educational materials is so critical for patients, physicians, and health plans as well. We hope that the FDA will stand by its public commitment to release more reading materials, more videos, more web info, etc. It is especially important at this juncture; we are seeing misinformation and a lack of clarity on certain things.
IS THE BIOSIMILAR ACTION PLAN ACTIONABLE?
BR&R: One of the biggest barriers to biosimilar access is the patent thickets. The rebate trap problem is another story. What power does HHS have to clear out the patent thickets? Or is this an area that can only be addressed by Congress?
Burich: This is the most difficult part of the Action Plan, because it is unclear who can truly implement change and what change might be realistic. We have to protect true innovation that’s important to all stakeholders.
At the same time, there’s no question that patent litigation is the leading barrier to biosimilar accesss. Some makers of branded pharmaceuticals have constructed patent thickets so that they could sustain prolonged, expensive litigation against competitors, while stifling competition. Humira is the prime example: More than 15 years after the molecule was approved , no biosimilar is being marketed – in the U.S. What the answer is and which government agency can effect change has yet to be determined.
BR&R: That change won’t come quickly, in any case. Whether enacted by Congress or the Office of the Inspector General, which may have to reinterpret the safe harbor statutes, this may only first apply to the second-generation of biosimilar agents, beyond 2021 perhaps. It seems likely that this will be a very deliberate process.
Burich: I do believe Commissioner Gottlieb is thinking about both how to get more products launched in the short term and also the long-term vision of a sustainable biosimilar market. That is such an important part of the problem.
We were very happy that the FDA had their public hearing. The FDA panel asked a lot of thoughtful and probing questions to the individual speakers. We are fully supportive of the Action Plan and its individual components. If we saw all of those things come together and start to see action, including finalizing the interchangeability guidance and providing more education, the biosimilar market would be in a far better place.
BR&R: We say that biosimilar manufacturers can make their products attractive to payers, but payers need to play a positive role here. Commissioner Gottlieb has said that payers have to help in this process by taking the long-term view, by not automatically sticking with the reference product because of the rebate revenue. They have to be open to using the biosimilars and nurturing the health of the industry. Is there anything else the biosimilar manufacturer can do to convince payers to make this market viable?
Burich: Certainly, biosimilar manufacturers have to approach these payer negotiations and conversations with competitive and innovative contracting approaches. That does not just include pricing but also how do you drive volume and true savings to both payers and patients. That kind of innovative approach is necessary, because we know it’s a challenging market.
Biosimilar manufacturers have to look at the whole picture as well. That means providing targeted patient/physician services to really help ensure that the switching experience is seamless for the patient and the physician so that biosimilar utilization is not viewed as something very disruptive.
In the first portion of a two-part interview with Molly Burich, MS, Director, Public Policy: Biosimilars and Pipeline, Boehringer Ingelheim, we cover the challenges of driving biosimilar uptake, as well as the unique situation that has focused this manufacturer’s attention on biosimilars and interchangeability.
BR&R: The viability of the US biosimilar industry is being challenged if companies cannot rely on revenue from switching, especially for the autoimmune category.
Molly Burich, MS
Molly Burich: Yes, biosimilar uptake is certainly going to be dependent on switching. But switching comes in a few different types. One case involves patients who are going to be switched to a therapy with a different mechanism of action. Perhaps their existing therapy no longer works (or didn’t work in the first place).
Another case is medication substitution by the physician. The doctor drives that decision to switch the patient either to a biosimilar or to an interchangeable.
Lastly is automatic substitution, which will come as a result of interchangeability and enabled by state laws. However, that is only in play once a product gains the interchangeability designation.
All of those are important components, but certainly switching overall is an important part of the market viability.
BR&R: When we’re talking about automatic switching, multiple stakeholders are involved, including the prescribers, pharmacies, payers, patients. And none of it matters if we don’t have an interchangeable product or even final guidelines from the FDA on interchangeability. In retrospect, should we have made automatic switching for biosimilars based on something other than interchangeability?
Burich: There are a lot of stakeholders involved and this is. why multiple ways of switching will likely occur. In terms of switching, interchangeability allows pharmacists to switch one reference product prescription for an interchangeable one without intervention of the physician at the front end—pending state laws of course. The physician must be notified of the change.
In our opinion though, automatic switching is certainly not the only way to drive uptake of biosimilars. We believe physician-driven switching and payer-drive substitution via formulary decision-making are very important to drive the uptake of biosimilars.
BOEHRINGER INGELHEIM’S SINGULAR PRODUCT FOCUS
BR&R: Biosimilar utilization, and the overall market, has been growing slowly since the first biosimilar approval. Prospective biosimilar manufacturers have tended to jump into the market with both feet, filling their pipelines with multiple biosimilar agents. Boehringer Ingelheim may be the only major manufacturer with a single biosimilar listed on its pipeline web page. Is the company in a wait-and-see mode, to see if the industry will survive? Or is Boehringer making further investments in biosimilar development behind the scenes?
Burich: We are constantly in an evaluation process of our portfolio. Obviously, we are focused on our approved biosimilar Cyltezo® (adalimumab-adbm) and also on interchangeability, here in the U.S. That is our focus area. We believe that the introduction of biosimilars will improve the lives of patients, as well as contribute to the quality and economic sustainability of healthcare systems.
INTERCHANGEABILITY: MISUNDERSTOOD BUT NO SILVER BULLET
BR&R: The issues around interchangeability are particularly frustrating. At the time the BPCIA was written, was the concept of interchangeability (which does not exist in EMA regulations) an attempt to give prescribers and consumers a warm and fuzzy feeling of an AB-rated generic?
Burich: It’s an important question. As you said, when the BPCIA was written, interchangeability was viewed as a sort of silver bullet. The reality is that interchangeability is an important concept, but perhaps it makes more sense for only certain products. As we gain experience in the biosimilar market, we’re starting to see this.
We believe in the concept of interchangeability and in what the FDA has put forth about interchangeability. We do think there are questions about how an interchangeable product may be perceived compared with one that is not interchangeable. In our comments to the FDA, we encouraged the FDA to come out with educational materials that are geared toward talking about interchangeability, and talking about switching. These are all important questions and need to be addressed for the broad stakeholder community. The FDA is obviously best positioned to bring that type of education in the next round of materials they develop.
BR&R: We’ve heard a great deal about people mischaracterizing interchangeable products as being superior to biosimilars (for the same reference product). Why is this differentiation so important?
Burich: This issue speaks to education. All people engaging in the biosimilar space must realize that the designation of interchangeability does not mean it’s a higher-quality, safer, or more-efficacious product. It means that the manufacturer has conducted additional studies required by the FDA to enable that automatic substitution at the pharmacy level.
The FDA has issued clarifying pieces of information and education on their website about this, but there is room for more. The reality is that when a drug is approved as a biosimilar, it has attained the foundational designation proving that the drug is highly similar to the reference biologic, without any clinically meaningful differences. On the other hand, gaining the interchangeability designation is about conducting trials of multiple switches within the patient and expecting the same results in any given to patient. Those are two different distinctions. It proves something different, allowing for automatic substitution to occur.
In part two and the conclusion of this interview, which will be published in a separate post, Molly Burich speaks to Boehringer Ingelheim’s progress in Cytelzo’s interchangeability studies, its plans for the product in Europe in the face of several adalimumab biosimilars launches in the EU, and also the complexity inherent in CMS’s plans to move biologic agents from part B to part D coverage.
The Biologics and Biosimilars Collective Intelligence Consortium (BBCIC) is the organization that will perform the critical task of postmarketing surveillance for biosimilars and their reference biologics. Started in 2015, the BBCIC is a nonprofit, scientific public service initiative, which partners with multiple stakeholders to accomplish its mission. In the conclusion of our two-part interview with Cate Lockhart, PharmD, PhD, we explore how BBCIC is communicating with its stakeholders and whether the FDA’s four-letter suffix for biosimilars will assist in its tracking efforts.
GETTING UP TO DATE WITH BBCIC ACTIVITIES
BR&R: Let’s talk a bit about what BBCIC is doing today. Can you give me an update on current BBCIC programs?
LOCKHART: Sure. We have completed our first round of descriptive analyses, composed of four studies that were designed to (1) characterize our patient population for these specific disease states and (2) understand our data. Can we reliably identify the cohorts of patients we’re interested in studying? Can we identify the outcomes that we’re interested in reliably? Do we trust our results, based on existing literature? What are the gaps in our capabilities? Those initial studies are completed, and we’re in the process of publishing those findings.
Cate Lockhart, PharmD, PhD
In the meantime, we have four work groups that are focused on methodological issues or filling in some of our data gaps. One group is working on best practices in comparative-effectiveness research (CER) methods. A lot of people are doing large-scale CER like this, but little consensus exists on best practices for performing it. We’ll come up with our own recommendations that will help guide our future research.
Another work group is specifically looking at switching patterns. The first phase of that work is completed. They identified methodological considerations for best approaching switching in the context of our research. We’ll soon be moving into the second phase, where we dig into BBCIC data and do a descriptive analysis, then we’ll start to understand the switching patterns in anti-inflammatory conditions, especially rheumatoid arthritis, and Crohn’s disease or ulcerative colitis.
BR&R: Will you be looking at switches from one biologic to another biologic? Or from one infliximab biosimilar to another?
LOCKHART: Well, that’s a good question. With rheumatoid arthritis, patients may change therapies many times and with different types of treatments. So we’re really looking at any and all of those. We are starting to get enough utilization of infliximab biosimilar products that we’re hopeful that we can start to include them in the analysis.
BR&R: When might we read some published data on some real-world evidence with biosimilars? Will it be in early 2019?
LOCKHART: It will take a bit longer. As everyone in the US has experienced, biosimilar uptake and utilization has been slower than expected. As we discussed before, in order to do robust research in this space, where we are looking at relatively rare outcomes—whether it is safety or effectiveness outcomes—we need enough utilization numbers in order to start that research.
We are in the process of initiating our first CER study in G-CSF products, because we believe we do have enough biosimilar utilization in that category to begin.
We have one project that’s looking at NDC and J-codes to evaluate how physician offices are coding their utilization of and administration of both the biosimilar products and their reference biologics. We’re just in the data analysis phase of that, so we do expect to see some publications coming out of that in 2019. We will be presenting five posters at AMCP Nexus in late October.
BR&R: One of the missions of BBCIC is to be transparent about what it is doing. Please tell me about the communication efforts of the organization.
LOCKHART: One of major efforts this year is to get the word out about BBCIC; too few people are aware of the work being done.
Part of that is because during the first couple of years, BBCIC, which was officially convened in 2015, was very much in a start-up phase. The organization was just getting off the ground and running. My role is to begin the transition from start-up phase into one with much more of a public face.
We’ve moved beyond this start-up phase. We’ve finished research projects. We’re starting to publish based on these projects. Personally, I’m accepting any speaking opportunity I can. I’m encouraging BBCIC participants to go to meetings and present. We are starting to get a little bit more traction in that respect.
Another effort is our quarterly newsletter, which very cleverly is called the BBCIC Quarterly. I’m producing the newsletter as a vehicle to keep the broader community—and our current participants—apprised of where we are in our research, our progress, our plans, and where we are publishing or presenting results. That newsletter is posted on BBCIC.org. Anybody who is interested in receiving the newsletter can contact me, and I’ll add them to the distribution list. I’m getting good feedback on it as a way to just keep people updated on the general goings-on.
BR&R: The outreach is really needed. During the course of FDA Advisory Committee meetings or conferences encouraging public comment, I’ve heard many times the mantra “we need to track the outcomes of these biosimilars” or “are we going to be doing postmarketing surveillance?” This gives the impression that there are no efforts underway to do just that.
THE FDA’S SUFFIX: FOUR LETTERS OR A FOUR-LETTER WORD?
Yet, BBCIC is focused on postmarketing surveillance. Now, part of the ability to conduct studies of biosimilars and their reference biologics involves another highly debated issue: the four-letter suffix. What is your opinion? Do we need four-letter suffixes of biosimilars and their reference biologics to track them?
LOCKHART: I see the value on both sides of this polarized debate. We’re doing our NDC and J-code exercise to look at how these drugs are ordinarily coded and whether the suffix is used. The reality is, usage is quite variable today.
On one hand, I agree that putting a suffix just on the biosimilar does put up a flag in people’s minds that there is something different about this product. We don’t do that with generic drugs, but certainly that there is a very different context between generic drugs and biosimilars. But the philosophy behind biosimilars and generics is really not that different.
For tracking purposes, there could be benefit in using the 4-letter suffix, but we can’t track it effectively if people are not using it for coding. And some of the coding systems being used today are not really designed to enter the suffix, so prescribers and administrative folks actually can’t code it in. There are some infrastructure challenges around that.
As much as I enjoy it when the government tells us to do things that are devoid of meaning, I personally believe that the use of random letters [in the suffix] makes it more confusing. I don’t remember what they are; the random suffixes are hard to remember, like –abda or -qbtx, with no meaningful context.
BR&R: At the recent AAM meeting, I heard Hillel Cohen from Sandoz say that of 69 adverse drug reports for filgrastim since 2015, all but four were filed without the four-letter suffix, and they were able to identify the correct brand, whether it was Zarxio®, Granix®, or Neupogen®.
It could be just as you said, that the administration system couldn’t handle the four-letter suffix, which is why it’s not entered. Or the providers ignored the four-letter suffix and used the INN and/or NDC code. Regardless, there doesn’t seem to be a rush to use the suffix in our early experience. Cate, from BBCIC’s perspective, is the suffix going to matter in terms of being able to track the use of a particular biosimilar?
LOCKHART: It’s really hard to predict. We’re in the position right now where infliximab is the only product where there’s more than one biosimilar on the market. If we’re looking at G-CSF agents, it’s not so complicated. It’s a hard question to answer, because it does rely so much on the infrastructure that we’re working with.
If we have these suffixes and nobody is using them, that’s missing the point. Some other countries don’t even bother with the suffixes—they just fill prescriptions with the brand name of the biosimilar. We still have a lot of inertia to overcome. Some of the controversy needs to be settled before we can even address the infrastructure problems.
From a BBCIC standpoint, I don’t think we know yet whether the use of the suffix will be critical, helpful, or neither. But, if providers or coders are not using the suffix in their claims, then it’s not helpful to us.
BR&R: Right. Well, it seems like pegfilgrastim may be the next test. We may have a second pegfilgrastim approval on or by November 3. That will mean two drug categories will have at least two biosimilars marketed. I’m guessing then it won’t be until into 2019 before we get a handle on who is using the four-letter suffixes for pegfilgrastim and who is not.
And the question of whether interchangeable products will get a unique identifier, no suffix, or something similar to the random four-letter suffix is still unanswered.
LOCKHART: That’s right. We still don’t know.
See Part 1 of our interview with Dr. Lockhart by clicking here.
A German manufacturer is considering its options after the successful completion of two clinical studies involving a pegfilgrastim biosimilar (MSB11455).
Fresenius Kabi, which completed its purchase of the biosimilar business from Merck KGaA in September 2017, announced its investigational biosimilar agent had proved sufficiently similar to the reference product Neulasta® in these phase 1 investigations (conducted in healthy participants). These may serve as pivotal investigations for the manufacturer, which said in its release, “Both studies are designed to enable the application for marketing authorization in the EU and US.” This may be the first indication that Fresenius Kabi seeks to be a player in the US.
Fresenius Kabi does not yet have an approved biosimilar on the European market. It hopes that MSB11455 may propel its fortunes on both sides of the Atlantic.
In its first study, the company reported that its biosimilar “met all primary pharmacokinetic endpoints, [maximum plasma concentration], and area under the curve, as well as the primary pharmacodynamic endpoints of absolute neutrophil count (ANC).” Fresenius Kabi added that there were no meaningful differences in the frequency of adverse events in these healthy volunteers. The second study focused on the biosimilar’s potential for immunogenicity, and this was also determined to be no different between the reference drug and the biosimilar. In addition, neutralizng antibodies were not found.
If Fresenius Kabi proceeds with an application for approval in either market, it will find a good deal of competition for pegfilgrastim biosimilars. In Europe, up to 5 biosimilars may be approved (2 already are). In the US, Mylan’s product is the only one to be approved, but another (Coherus Biosciences) is expecting a decision from the Food and Drug Administration (FDA) in early November. Two others (Sandoz and Apotex) are seeking US drug approval.
In other biosimilar news…The Food and Drug Administration’s Oncology Drug Advisory Committee voted unanimously (16-0) today to recommend Celltrion’s CT-P10 rituximab biosimilar for approval. If the biosimilar is approved by the FDA, it will be marketed by Teva….Mundipharma purchased European biosimilar maker Cinfa, which has a pegfilgrastim that has received a CHMP recommendation for approval in the EU.
The Biologics and Biosimilars Collective Intelligence Consortium (BBCIC) is the organization that will perform the critical task of postmarketing surveillance for biosimilars and their reference biologics. Started in 2015, the BBCIC is a nonprofit, scientific public service initiative, which partners with multiple stakeholders to accomplish its mission. In the first post of our two-part interview with Cate Lockhart, PharmD, PhD, we explore how the organization is moving beyond start-up mode and towards providing real-world evidence on the efficacy and safety of biosimilars.
BR&R: Cate, what experiences or interests brought you to the BBCIC?
Cate Lockhart, PharmD, PhD
CATE LOCKHART, PHARMD, PHD: I was working in a small biotech company in Seattle. I was the health economics department. As is often the case in small biotech companies, it was a department of one.
I came into this role at the BBCIC in January 2018. Prior to that I’d been working on a part-time consultant basis with my predecessor, Bernadette Eichelberger, who was the architect of this organization. I had been working with her for a year and one-half or so, and to some extent, I learned about the inner workings of the BBCIC before I jumped into this role.
But what was really attractive to me was that it bridges a lot of areas of research, reflecting a lot of my experience in pharmaceutics, health economics, and pharmacy. This was, in so many ways, the perfect job for me because it really does marry all of those elements.
WHAT MAKES THE BBCIC TICK?
BR&R: Can you give us a little bit of a background on the collective intelligence methodology that underlies BBCIC.
LOCKHART: Sure. One of the unique elements of the BBCIC is our governance structure and the framework under which we operate. It’s a consortium, which means we bring together multiple stakeholders from a variety of perspectives. We have organizations who are manufacturers, we have managed care organizations, we have payers, we have some other nonprofit and some patient-engagement organizations. All of whom come to the table together for the common goal of conducting this research into the safety and effectiveness of biosimilars and their reference biologics.
Part of the logic behind that is, first, it helps to reduce some of the bias or perceived bias inherent in some research. Our work is not beholden to any one company, because all of the resources are pooled. In fact, some of our participating organizations are direct competitors, all sitting at the same table. That helps increase the credibility of the research.
Second, it gives us the opportunity to draw on the very broad expertise of our participants to hopefully make the research questions and the results that come out of our work relevant to as broad an audience as possible.
Third, it maintains a very high level of transparency. We operate under a charter. We have a board of managing directors. We also have a planning board or steering committee. Each of our participating organizations has a seat on the planning board. We have a science committee to help drive the scientific direction. We have a communications committee, and then we have our research teams. Each of these roles is clearly delineated in our charter, which is a publicly available document. We don’t operate in a black box.
BR&R: The BBCIC’s process is based on an existing program—the FDA’s Sentinel program.
LOCKHART: That’s a good point. The BBCIC is not part of the Sentinel program, but we leverage the infrastructure that was developed for Sentinel. The FDA spent a lot of time and money developing their common data model and coordinating its data partners to display and report their data in a specific way, so we’re leveraging that. There was no need to reinvent the wheel.
One key difference, in my opinion, is although our capabilities are very similar to Sentinel’s, our mission is a bit different. For one thing, we really are focused on biosimilars and their reference biologics. In that way, it fills a gap that FDA simply doesn’t have the bandwidth to address.
The research that BBCIC does is proactive. The FDA tends to be reactive, investigating potential safety signals once they occur. That is, they may receive a signal through their safety reporting system, at which time they can activate Sentinel and investigate the safety signal further. Their primary purview is response to safety.
On the other hand, BBCIC is proactively looking at safety and effectiveness in a real-world setting. That differentiates us to some extent from the other existing organizations that are doing this sort of large-scale, multi-stakeholder, multi-data source, observational research.
CAN WE CAPTURE SIGNALS OF WANING EFFECTIVENESS?
BR&R: Monitoring biosimilars’ efficacy or effectiveness seems to be extremely important, particularly with all of the discussions that we’ve heard about lot-to-lot variations and the potential for manufacturing changes. This also applies to the reference biologics.
Can you provide an example, if you can, of how the process might actually identify efficacy changes or maybe decreasing potency?
LOCKHART: We’re going into this under the assumption that there will be no discernible difference, because that’s what we believe. These products have gone through the FDA review process, and we trust the FDA reviewers and their process.
Effectiveness is a challenging outcome to measure when you’re analyzing administrative claims alone. So to get a better picture, we may be interested in looking at different types of data sources. Part of our work is to understand those gaps in data to measure effectiveness and identify other data sources that can help fill in the picture.
Because of the scale of our data “network,” for lack of a better word, we can more easily identify rare events. We expect the safety signals or signs of effectiveness changes to be [hiding] in the data, so you need to have a very broad, diverse network of data to identify those signals. With BBCIC, we have that breadth of administrative claims data to help answer some of those questions.
BR&R: When using an administrative data set, can we somehow identify switching as a marker for waning effectiveness? Hypothetically, if patients with rheumatoid arthritis who were receiving a biosimilar for etanercept began switching more frequently over time to a biosimilar or reference biologic for adalimumab, for instance, could that potentially hint at decreasing effectiveness? I’m guessing that one would need a tremendous amount of data collected from the various participating institutions to validate that sort of trend.
LOCKHART: It’s true. Switching is an interesting topic, and we’ve devoted a fair bit of time to it. When you’re starting to do observational research with administrative claims, adding switching into the mix introduces some difficulties in analysis.
You’re right—you do need a lot of data in order to really evaluate any switching trends, and you need enough longitudinal data in order to evaluate any outcomes. When a patient switches from one biologic to a non-biologic, from one biologic to another with a different mechanism of action, or between a biosimilar and reference product, you need to have data covering sufficient time of exposure in order to identify an emerging signal. We’re looking into some of those questions today.
It’s hard to answer the question of why a patient switched therapies. It could be a clearly administrative reason: A certain payer made a drug formulary change and suddenly covers only the biosimilar and not the reference product. That’s easier to identify. In some diseases, like rheumatoid arthritis, patients are on a pretty circuitous trajectory anyway, and they do a lot of switching. It’s hard to tease out what outcomes are a result of a switch versus a result of the underlying disease. Those are some of the difficult questions that we have to wrap our heads around.
We’re also looking into some methodological questions and developing some best practices for how to examine switching, because it does introduce quite a bit of complexity. That said, the European experience has yielded a richer field of data because of their longer utilization of biosimilars. Their studies have not shown, in the literature I’ve seen, any differences in safety or efficacy based upon switching. But again, teasing out the nuances is challenging with any data source.
Part 2 and the conclusion of our interview with Dr. Lockhart will be published in an upcoming post.

 The Center for Biosimilars reported an Email exchange with the Danish national tendering authority Amgros, which manages the country’s bidding system. Amgros confirmed that Abbvie did not provide the best bid for two tenders for adalimumab (covering January to March 2019 and covering April to December 2019). Five companies (including Abbvie) competed for the national tenders. Although Abbvie did not rank best for pricing, agreements were signed with all five companies.
The Center for Biosimilars reported an Email exchange with the Danish national tendering authority Amgros, which manages the country’s bidding system. Amgros confirmed that Abbvie did not provide the best bid for two tenders for adalimumab (covering January to March 2019 and covering April to December 2019). Five companies (including Abbvie) competed for the national tenders. Although Abbvie did not rank best for pricing, agreements were signed with all five companies.


 In its first study, the company reported that its biosimilar “met all primary pharmacokinetic endpoints, [maximum plasma concentration], and area under the curve, as well as the primary pharmacodynamic endpoints of absolute neutrophil count (ANC).” Fresenius Kabi added that there were no meaningful differences in the frequency of adverse events in these healthy volunteers. The second study focused on the biosimilar’s potential for immunogenicity, and this was also determined to be no different between the reference drug and the biosimilar. In addition, neutralizng antibodies were not found.
In its first study, the company reported that its biosimilar “met all primary pharmacokinetic endpoints, [maximum plasma concentration], and area under the curve, as well as the primary pharmacodynamic endpoints of absolute neutrophil count (ANC).” Fresenius Kabi added that there were no meaningful differences in the frequency of adverse events in these healthy volunteers. The second study focused on the biosimilar’s potential for immunogenicity, and this was also determined to be no different between the reference drug and the biosimilar. In addition, neutralizng antibodies were not found.
 The research that BBCIC does is proactive. The FDA tends to be reactive, investigating potential safety signals once they occur. That is, they may receive a signal through their safety reporting system, at which time they can activate Sentinel and investigate the safety signal further. Their primary purview is response to safety.
The research that BBCIC does is proactive. The FDA tends to be reactive, investigating potential safety signals once they occur. That is, they may receive a signal through their safety reporting system, at which time they can activate Sentinel and investigate the safety signal further. Their primary purview is response to safety.